УДК: 616-007-053.1
Год издания: 2012
Мутации гена MPZ как причина невральной амиотрофии Шарко-Мари-Тус тип 1В
Осадчук Т.В., Моссэ К.А., Румянцева Н.В.
Рубрики: 76.03.39
Республиканский научно-практический центр "Мать и дитя"
Тема НИР: «Разработать и внедрить технологию полного сканирования нуклеотидной последовательности ДНК генов, ответственных за развитие наследственных и врожденных заболеваний человека»
Сроки выполнения НИР: январь 2010 г. — декабрь 2012 г.
Научный руководитель: канд. мед. наук И.В. Наумчик
Источник финансирования: госбюджет.
Цель:определение мутаций гена MPZ, приводящих к невральной амиотрофии Шарко-Мари-Тус тип 1B (СМТ тип 1В).
Невральная амиотрофия Шарко-Мари-Тус (СМТ — от англ. Charcot-Marie-Tooth), или наследственная моторно-сенсорная невропатия (НМСН) — обширная группа генетически гетерогенных заболеваний, характеризующаяся симптомами прогрессирующей поли-невропатии с преимущественным поражением мышц дистальных отделов конечностей. НМСН является самой частой среди наследственных заболеваний перифе-рической нервной системы. Частота всех форм НМСН варьирует от 1 до 4 на 10000 в различных популяциях.
СМТ тип 1В является вторым по частоте среди аутосомно-доминантных форм СМТ после СМТ тип 1А и третьим по частоте с учетом Х-сцепленной формы СМТ тип 1Х. Ген MPZ (myelin protein zero), мутации в котором приводят к развитию СМТ тип 1В, локализован на хромосоме 1 в сегменте q22.1-q23, состоит из 6 экзонов и кодирует миелиновый белок Ро, который является основным структурным компонентом периферического миелина и играет важную роль в процессе укладки отдельных слоев миелиновой оболочки (ком-пактизации миелина).
Клинические проявления отдельных генетических вариантов НМСН имеют значительное сходство, что затрудняет диагностический этап медико-генетического консультирования и обуславливает необходимость проведения ДНК-анализа для дифференциальной диагностики различных форм.
На первом этапе диагностики в отобранной группе пациентов с клиническими симптомами моторно-сенсорной невропатии были исключены CMT тип 1A и CMT тип 1X. Для определения мутаций в гене MPZ ресеквенирование кодирующей последовательности гена проведено в 58 образцах ДНК (50 пробандов, 8 родственников). По имеющимся данным, большинство мутаций гена MPZ (около 77%) обнаружено во втором и третьем экзонах, поэтому поиск нуклеотидных замен выполнен в первую очередь именно в них.
Мутации во втором и третьем экзонах гена MPZ об-наружены в двух семьях у 5 человек: у 2 из 50 (4%) про-бандов и у 3 из 8 обследованных родственников. Характеристики нуклеотидных замен приведены в табл.
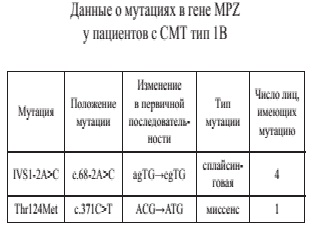
Одна из обнаруженных мутаций с.371C>T, описанная ранее различными группами исследователей, приводит к замене треонина на метионин в 124 кодоне (Thr124Met) и имеет разнообразные клинические проявления, включающие помимо клинической картины СМТ нейросенсорную тугоухость/глухоту, хронический кашель и синдром Аргайла–Робертсона. Вторая из обнаруженных мутаций описана нами впервые в мире и представлена однонуклеотидной заменой во второй позиции 3′-последовательности 1 интрона гена MPZ и обозначается как IVS1-2A>C (сплайсинговая). Данная мутация ассоциирована с тяжелой моторно-сенсорной невропатией: начало заболевания на первой декаде жизни, мышечная гипотония, нарушение походки, слабость мышц дистальных отделов конечностей, потеря чувствительности. Такая же нуклеотидная замена идентифицирована у трех родственников пробанда: у отца пробанда и двух ее сыновей в возрасте 2,5 года и 4 мес. Новая мутация не была обнаружена у здоровых членов семьи, а также в контрольной группе.
Обнаружение нуклеотидных замен в исследуемой группе пациентов свидетельствует о необходимости исследования данного гена с целью дифференциальной диагностики типа СМТ. Применение методов ДНК-анализа способствует не только раннему выявлению заболевания, но также позволяет своевременно выявлять бессимптомных носителей и проводить пренатальную диагностику в отягощенных семьях.
Область применения: медицинская генетика, неврология.
Предложения по сотрудничеству: консультативная и диагностическая помощь пациентам неврологических отделений, центров медико-генетического консультирования.